Ускорение предсказания структуры белков на уровне протеома
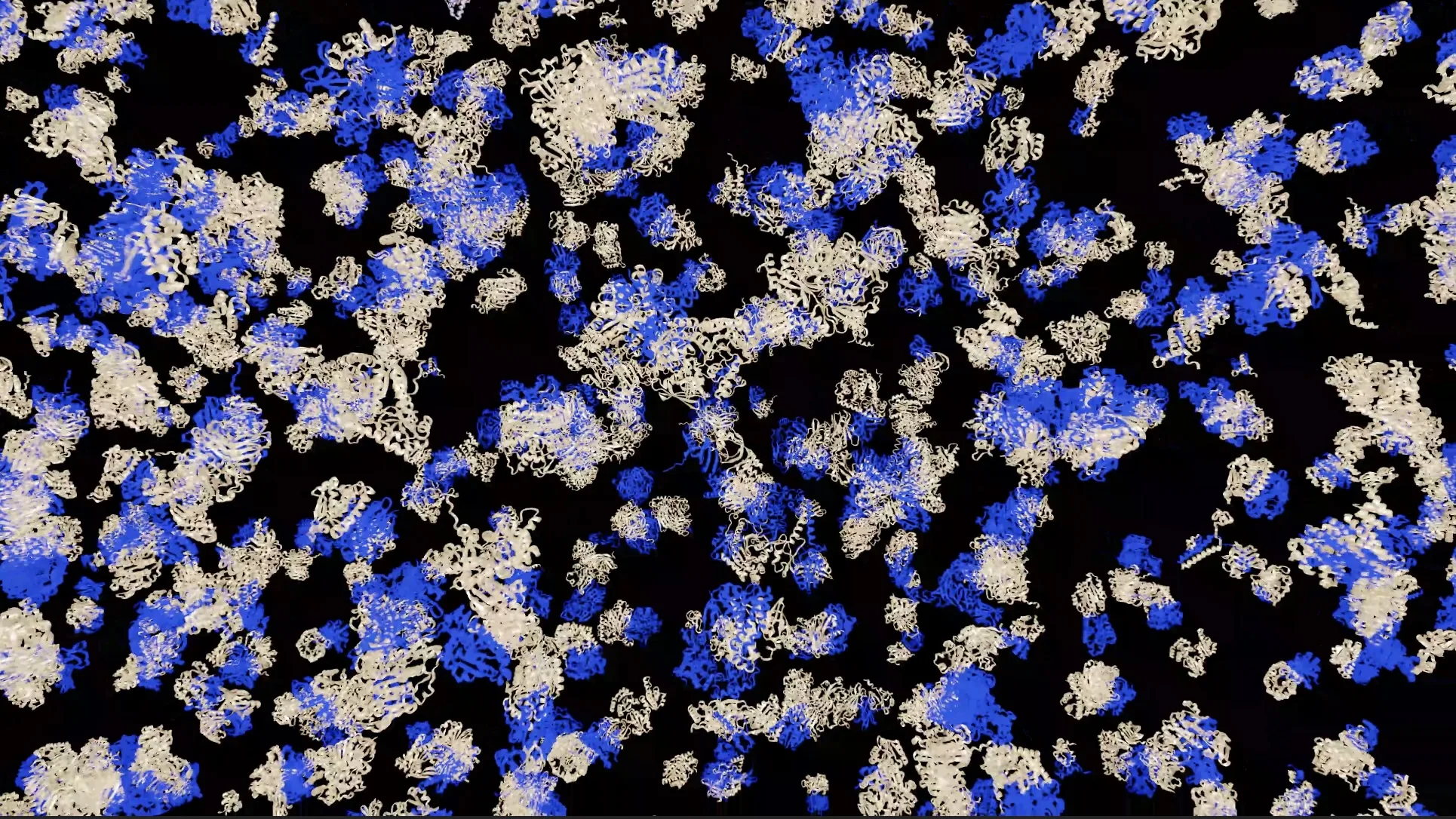
Белки редко функционируют в изоляции как отдельные мономеры. Большинство биологических процессов управляются взаимодействиями между белками, образующими белковые комплексы, структуры которых описываются в иерархии белковой структуры как четвертичные представления. Это представляет собой один уровень сложности выше третичных представлений, трехмерных структур мономеров, которые стали известны с появлением AlphaFold2 и созданием Базы данных белковых структур. Однако структурная информация для подавляющего большинства комплексов остается недоступной.
База данных структур белков AlphaFold (AFDB), совместно разработанная Google DeepMind и Европейским институтом биоинформатики (EMBL-EBI), преобразила доступ к мономерным белковым структурам, но взаимодействие с учетом структуры на уровне протеома по-прежнему остается узким местом с уникальными проблемами. К ним относятся огромное комбинаторное пространство взаимодействий, высокая вычислительная стоимость генерации множественных последовательных выравниваний (MSA) и сворачивания белков, а также проблемы с масштабированием вывода для миллионов комплексов.
В недавней работе мы расширили AFDB с помощью крупномасштабных предсказаний гомомерных белковых комплексов, созданных с помощью высокопроизводительного конвейера на основе AlphaFold-Multimer, что стало возможным благодаря ускоренным вычислениям от NVIDIA. Мы также предсказали гетеромерные комплексы для сравнения точности различных модальностей предсказания комплексов. В частности, для предсказаний этих наборов данных мы использовали ускорения на уровне ядра от MMseqs2-GPU для генерации MSA и NVIDIA TensorRT для сворачивания белков на основе глубокого обучения.
Данная статья описывает основные принципы, которые мы использовали для увеличения пропускной способности сворачивания белков, начиная с использования библиотек и SDK до оптимизаций, снижающих вычислительную сложность нагрузки. Эти принципы могут помочь вам настроить аналогичный конвейер, заимствовав техники, которые мы использовали для создания этого нового набора данных.
Если вы являетесь вычислительным биологом, исследователем ИИ, инженером HPC или биоинформатиком, вы узнаете, как разработать стратегию предсказания комплексов на уровне протеома, отделить генерацию MSA от предсказания структуры для повышения эффективности и масштабировать рабочие процессы AlphaFold-Multimer на кластерах GPU.
Amazon Bedrock внедряет новые возможности для взаимодействия с агентами
Как работают модели визуально-языкового действия (VLA) для роботов
Похожие статьи
Будущее сжатия данных: от пикселей до ДНК
Будущее сжатия данных охватывает все типы информации, от геномов до видео, расширяя возможности цифровых технологий.

Сравнение ИИ между США и Китаем: разрыв в ответственности растёт
Отчет Stanford University показывает, что разрыв в производительности ИИ между США и Китаем закрылся, но проблемы с ответственностью и безопасностью остаются.
MIT создает многозадачные квантовые сенсоры для одновременного измерения
MIT разработали квантовые сенсоры, способные одновременно измерять несколько физических величин.