Accelerating Protein Structure Prediction at Proteome-Scale
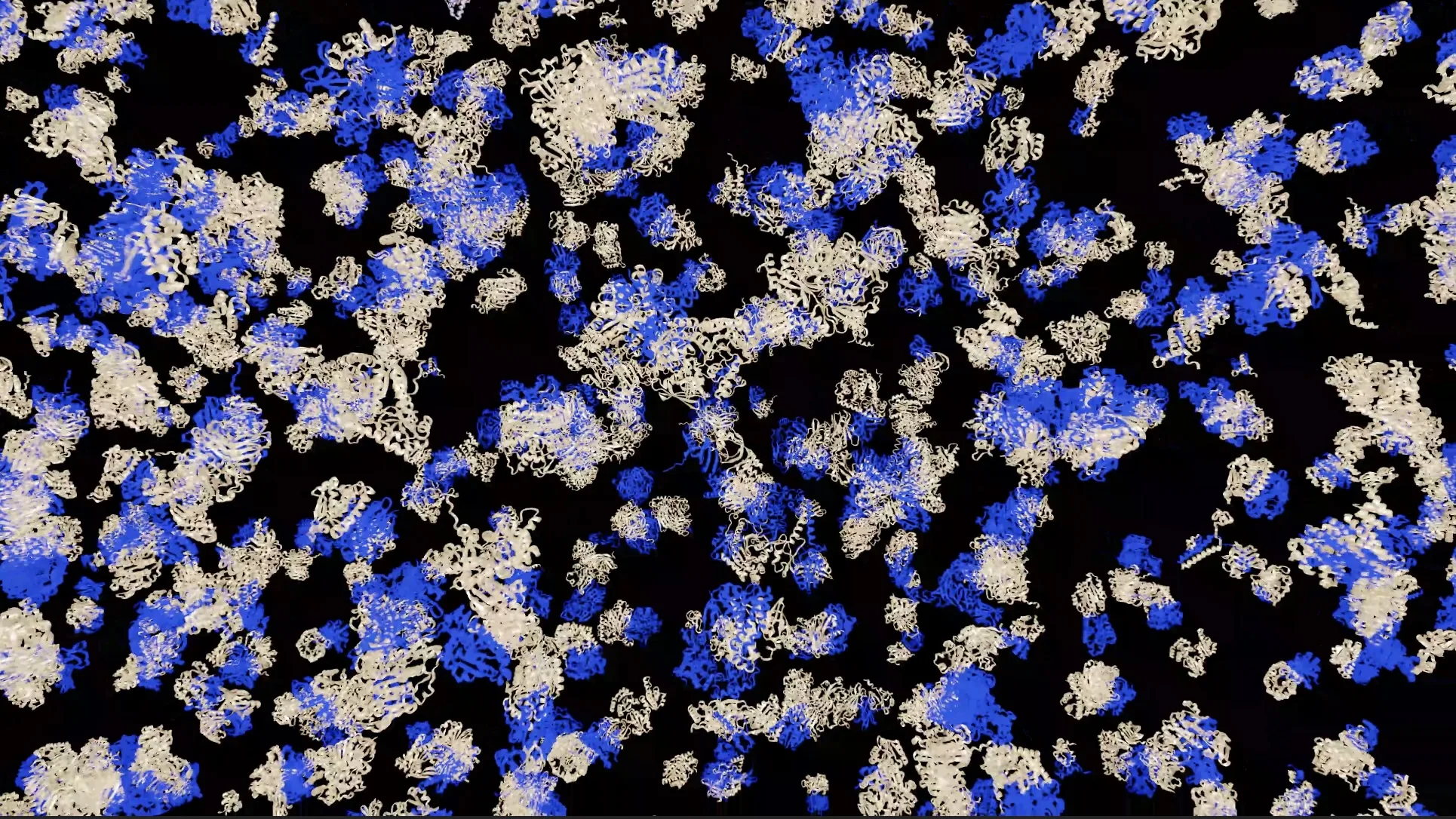
Proteins rarely function in isolation as individual monomers. Most biological processes are governed by proteins interacting with other proteins, forming protein complexes whose structures are described in the hierarchy of protein structure as the quaternary representation. This represents one level of complexity up from tertiary representations, the 3D structure of monomers, which are commonly known since the emergence of AlphaFold2 and the creation of the Protein Data Bank. However, structural information for the vast majority of complexes remains unavailable.
The AlphaFold Protein Structure Database (AFDB), jointly developed by Google DeepMind and EMBL’s European Bioinformatics Institute (EMBL-EBI), transformed access to monomeric protein structures, but interaction-aware structural biology at the proteome scale has remained a bottleneck with unique challenges. These include massive combinatorial interaction space, high computational cost for multiple sequence alignment (MSA) generation, and protein folding, as well as inference scaling across millions of complexes.
In recent work, we extended the AFDB with large-scale predictions of homomeric protein complexes generated by a high-throughput pipeline based on AlphaFold-Multimer, made possible by NVIDIA accelerated computing. We also predicted heteromeric complexes to compare the accuracy of different complex prediction modalities. For these predictions, we leveraged kernel-level accelerations from MMseqs2-GPU for MSA generation and NVIDIA TensorRT for deep-learning-based protein folding.
This article describes the major principles we adopted to increase protein folding throughput, from adopting libraries and SDKs to optimizations that reduce the computational complexity of the workload. These principles can help you set up a similar pipeline by borrowing from the techniques we used to create this new dataset.
If you are a computational biologist, AI researcher, HPC engineer, or bioinformatician, you will learn how to design a proteome-scale complex prediction strategy, separate MSA generation from structure inference for efficiency, and scale AlphaFold-Multimer workflows across GPU clusters.
Amazon Bedrock introduces new capabilities for agent interaction
How Visual-Language-Action (VLA) Models Work for Robots
Related articles
The Future of Data Compression: From Pixels to DNA
The future of data compression encompasses all types of information, from genomes to video, expanding the capabilities of digital technologies.

US-China AI performance gap closes, but responsibility gap widens
The Stanford University report indicates that while the US-China AI performance gap has closed, issues with responsibility and safety remain.
MIT develops multitasking quantum sensors for simultaneous measurements
MIT researchers have developed quantum sensors capable of measuring multiple physical quantities simultaneously.